AML-67-001 tutorial
Prepare the parameters
-D: AML_67_001-i: ReadCounts_AML_67_001.tsv (Input read count file name , present in the folder Count_file_and_gq_file-g: GQ_AML_67_001.tsv (Quality file name, present in the folder Count_file_and_gq_file-o: AML_67_001_ (Output prefix)
Running PHALCON
- The output obtained from MissioBio Tapestri pipeline is in the form of loom files. The loom file for AML-67-001 is present in the folder loom file.
- To convert the loom file in PHALCON-style input use the script
loompy_to_readcount_indels.pypresent in the same folder
Input : Pass the readcount file, genotype quality file and the output prefix name. Run the following command:
python Algorithm_phalcon_indels.py -D AML_67_001 -i ReadCounts_AML_67_001.tsv -g GQ_AML_67_001.tsv -o AML_67_001_
Output : You will get the variants inferred by PHALCON in form of a VCF file.
Subset vcf output:
##fileformat=VCFv4.1
##source=OurAlgoOurAlgo v0.1.0
##FILTER=<ID=LowQual,Description="Low quality">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth; some reads may have been filtered">
##FORMAT=<ID=AD,Number=.,Type=Integer,Description="Allelic depths for alt alleles">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth (reads with MQ=255 or with bad mates are filtered)">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=PL,Number=G,Type=Integer,Description="Normalized, Phred-scaled likelihoods for genotypes as defined in the VCF specification">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT cell1 cell2 cell3 cell4 cell5 cell6 cell7 cell8 cell9 cell10 cell11 cell12 cell13 cell14 cell15 >
chr1 115258748 * C T * PASS DP=809943 GT:AD:DP:GQ:PL 0/1:392:392:99 0/1:227:430:99 0/0:0:444:99 0/0:0:205:99 0/0:0:0:99 0/0:0:211:99 >
chr1 115258749 * T G * PASS DP=805084 GT:AD:DP:GQ:PL 0/1:392:392:99 0/1:227:430:99 0/0:0:0:99 0/0:0:207:99 0/0:0:104:99 0/0:0:209:99 >
chr2 25463285 * GCGGTAGAACTCAAA G * PASS DP=188342 GT:AD:DP:GQ:PL 0/1:0:97:99 0/1:6:23:99 0/1:0:0:12 0/1:26:44:99 0/0:0:124:99 0/1:47:4>
chr2 25469085 * CG C * PASS DP=216991 GT:AD:DP:GQ:PL 0/0:0:149:99 0/0:0:63:99 0/1:0:19:57 0/0:0:149:99 0/0:1:124:99 0/1:0:0:99 >
chr2 25469913 * C T * PASS DP=128427 GT:AD:DP:GQ:PL 0/1:0:39:99 0/1:39:60:99 0/1:32:32:95 0/1:22:33:99 0/1:66:93:99 0/1:29:33:35 >
chr2 198266943 * C T * PASS DP=689574 GT:AD:DP:GQ:PL 0/1:307:313:99 0/1:72:72:99 0/1:149:151:99 0/1:217:217:99 0/1:242:242:99 0/1:153:154:99 >
chr2 198267770 * G GAA * PASS DP=257605 GT:AD:DP:GQ:PL 0/1:46:92:99 0/1:0:0:99 0/1:27:59:99 0/1:15:22:99 0/1:56:117:99 0/1:0:0:99 >
chr4 55592244 * G A * PASS DP=266536 GT:AD:DP:GQ:PL 0/1:93:325:99 0/1:0:0:99 0/1:0:0:99 0/1:0:0:99 0/1:15:65:99 0/1:0:0:99
Post-processing using dbSNP
The post processing includes removing variants with no mutation across all cells and removing clonal variants present in the dbSNP database 📑
- The dbSNP.vcf is present at the link dbSNP_database
Follow the steps below to generate the final vcf file of post-processed variants:
- Download the folder post-processing in your local system
- Open termnal inside the folder and type
jupyter-notebook - Open the notebook Post-processing_AML-67-001.ipynb
- Run
dbsnp_for_aml_finite_sites.py - You will get a final list of variants in form of a vcf file in a file name ending with
_post_processed_final.vcf
NOTE: As input to the dbsnp_for_aml_finite_sites.py python file, you only need the vcf file you obtained after running PHALCON.
If required, change the location of the vcf file accordingly.
- Use
-ito provide location of the input vcf file - Use
-oto mention the name of the post processed vcf file
Annotate variants
For variant annotation using ANNOVAR, the necessary files and scripts are present in the annotating_variants link.
Use the following steps to annotate mutations:
- Download the folder annotating_variants in your local system
- Open termnal inside the folder and type
jupyter-notebook - Open the notebook Annotating_the_final_variants.ipynb
- Run
run_annovar_for_annotating_final_variants.py - You will get an annotated files of the final variants
NOTE: As input to the run_annovar_for_annotating_final_variants.py python file, you only need the post-processed vcf file.
If required, change the location of the vcf file accordingly.
- Use
-ito provide location of the post-processed input vcf file - Use
-oto mention the output tag of the annotated files
Visualization
The genotyped variants are shown in the form of a heatmap, and the chronological order of mutations is shown in the form of a phylogenetic tree.
The following files are needed to produce the final tree and genotypes (All of these files can be obtained by running the above pipelines):
-d: Tag for the dataset (OPTIONAL)(Acts as an identifier)--cluster_labels_file: Inferred cluster labels (Obtained as one of the files after running PHALCON)--lklhd_file: Likelihood file containing mutation likelihood for each cell and variant (Obtained as one of the files after running PHALCON)--final_df_file: Likelihood file containing mutation likelihood for each cell and variant (Obtained as one of the files after running PHALCON)--genotype_config_file: Genotype configuration file (Obtained as one of the files after running PHALCON)--newick_file: Newick format file (Obtained as one of the files after running PHALCON)--vcf_file: Final post-processed VCF file (Post-processed final VCF file, refer to Post-processing using dbSNP)--variant_function_file: Gene-annotated file of the post-processed VCF file, refer to Annotate variants)
Run the following command (all the files used in the command can also be found in the Input_for_visualisation folder for ease):
python phalcon_visualization_pipeline.py -d AML_67_001 --cluster_labels_file aml_67_001_inferred_cluster_labels.txt --lklhd_file aml_67_001_final_lklhds.tsv --final_df_file aml_67_001_final_df.tsv --genotype_config_file aml_67_001_Genotype_configuration.tsv --newick_file aml_67_001_inferred_tree.nw --vcf_file aml_67_001_post_processed_final.vcf --variant_function_file aml_67_001_output.avinput.variant_function
You will obtain the following outputs:
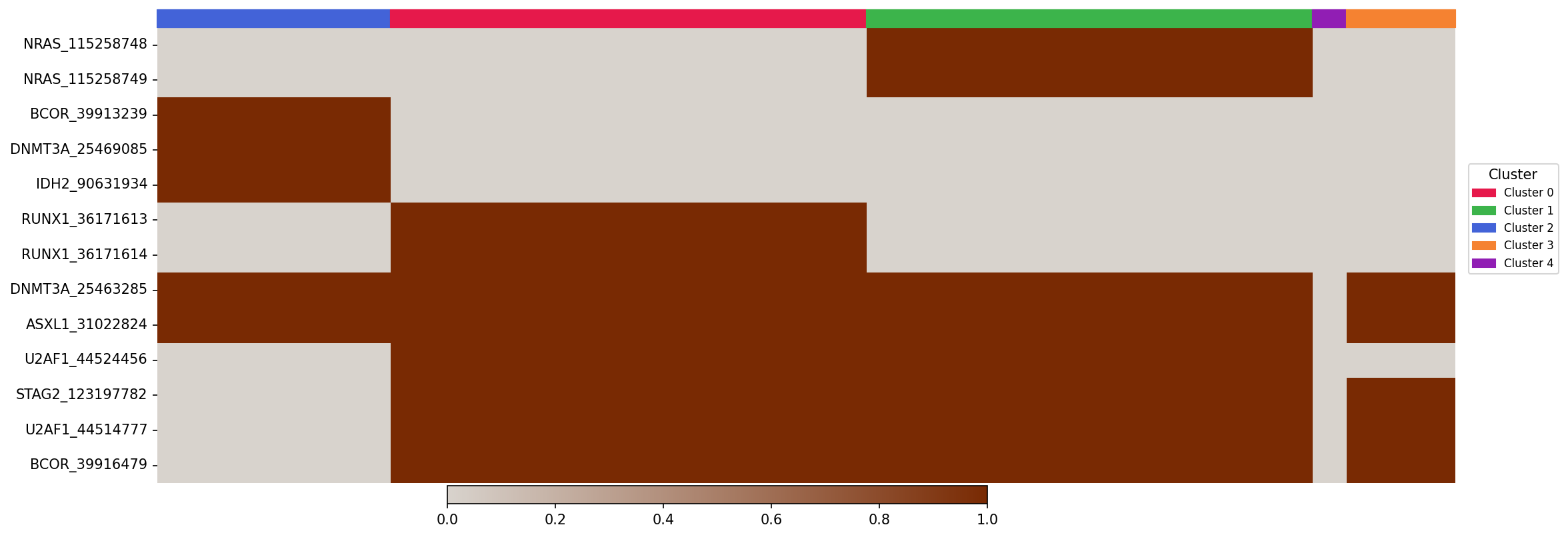
- Genotype heatmap:
The genotype heatmap shows the genotype profiles of each cell. There is a bar at the top which shows the cluster association of each cell.
- UMAP:
The umap shows the clusters obtained by PHALCON on a 2-d plane.

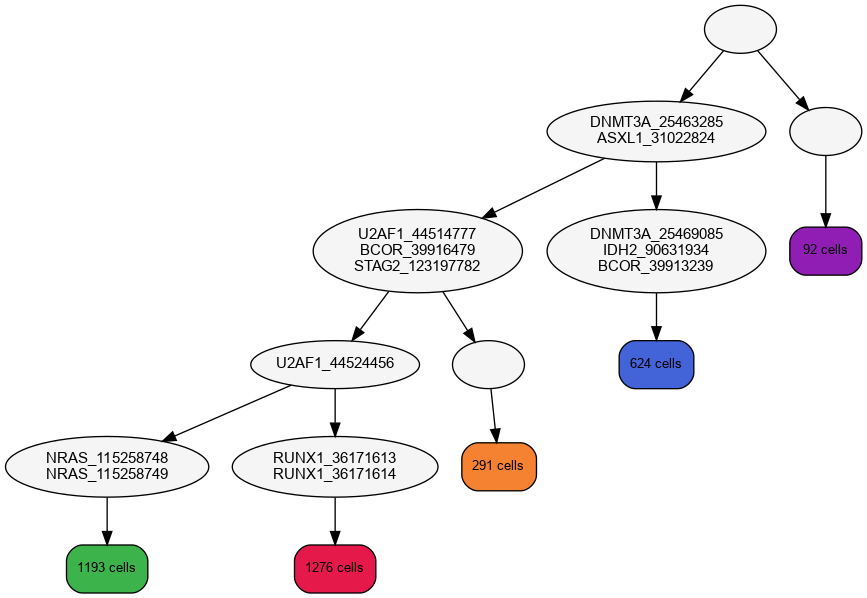
- Phylogenetic tree:
The reconstructed mutation history tree shows the order in which the mutations have appeared.
Co-occurence and mutual exclusivity patterns
Use the R script provided in the cooccurrence and mutual exclusivity script folder to produce the plot.
- Odds ratio plot:

The p-value for each association is present in form of a table as the output of the R script.